





Además de la tecnología, la síntesis de glucósidos siempre ha sido de interés para la ciencia, ya que es una reacción muy común en la naturaleza. Artículos recientes de Schmidt, Toshima y Tatsuta, así como numerosas referencias citadas en ellos, han analizado una amplia gama de potenciales sintéticos.
En la síntesis de glucósidos, un componente multiazúcar se combina con nucleófilos, como alcoholes, carbohidratos o proteínas. Si se requiere una reacción selectiva con uno de los grupos hidroxilo de un carbohidrato, todas las demás funciones deben protegerse en el primer paso. En principio, los procesos enzimáticos o microbianos, gracias a su selectividad, pueden reemplazar los complejos pasos químicos de protección y desprotección para la síntesis selectiva de glucósidos en ciertas regiones. Sin embargo, debido a la larga historia de los alquil glucósidos, la aplicación de enzimas en su síntesis no ha sido ampliamente estudiada ni aplicada.
Debido a la capacidad de los sistemas enzimáticos adecuados y los altos costos de producción, la síntesis enzimática de alquilpoliglucósidos no está lista para ser actualizada al nivel industrial y se prefieren los métodos químicos.
En 1870, MAcolley informó la síntesis de “acetoclorhidrosa” (1, figura 2) mediante la reacción de dextrosa (glucosa) con cloruro de acetilo, lo que eventualmente condujo a la historia de las rutas de síntesis de glucósidos.
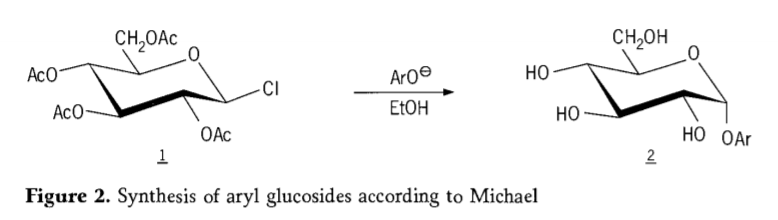
Posteriormente, se descubrió que los haluros de tetra-O-acetil-glucopiranosilo (acetohaloglucosas) eran intermediarios útiles para la síntesis estereoselectiva de glucósidos de alquilo puros. En 1879, Arthur Michael logró preparar glucósidos de arilo cristalizables a partir de los intermediarios de Colley y fenolatos (Aro-, Figura 2).
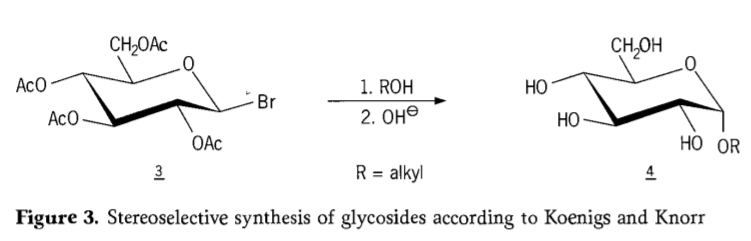
En 1901, la síntesis de Michael de una amplia gama de carbohidratos y agliconas hidroxílicas, cuando W. Koenigs y E. Knorr introdujeron su proceso mejorado de glicosidación estereoselectiva (Figura 3), se completó. La reacción implica una sustitución SN₂ en el carbono anomérico y se desarrolla estereoselectivamente con inversión de configuración, produciendo, por ejemplo, el α-glucósido 4 a partir del anómero β del intermedio aceobromoglucosa 3. La síntesis de Koenigs-Knorr se lleva a cabo en presencia de promotores de plata o mercurio.
En 1893, Emil Fischer propuso un enfoque fundamentalmente diferente para la síntesis de alquilglucósidos. Este proceso, conocido hoy como «glicosidación de Fischer», consiste en una reacción de glicosas con alcoholes, catalizada por ácido. Sin embargo, cualquier registro histórico debería incluir también el primer intento, descrito por A. Gautier en 1874, de convertir dextrosa con etanol anhidro en presencia de ácido clorhídrico. Debido a un análisis elemental engañoso, Gautier creyó haber obtenido una «diglucosa». Fischer demostró posteriormente que la «diglucosa» de Gautier era, en realidad, principalmente etilglucósido (Figura 4).
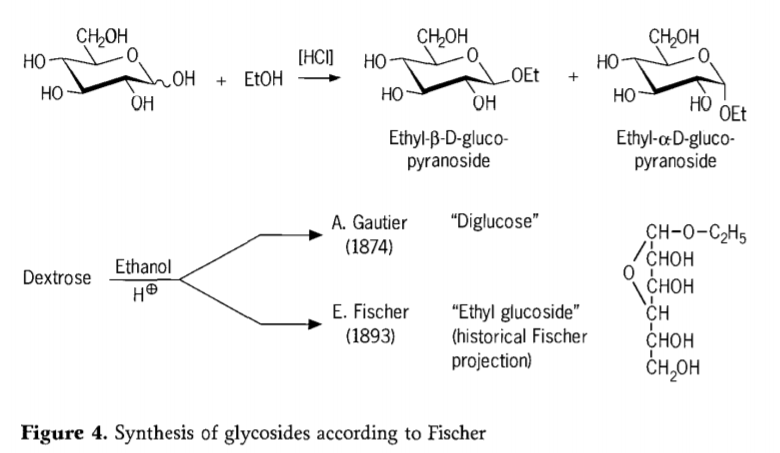
Fischer definió correctamente la estructura del etil glucósido, como se desprende de la fórmula furanosídica histórica propuesta. De hecho, los productos de la glucosidación de Fischer son mezclas complejas, en su mayoría en equilibrio, de anómeros α/β e isómeros de piranósido/furanósido, que también comprenden oligómeros de glucósido enlazados aleatoriamente.
Por consiguiente, no es fácil aislar especies moleculares individuales de las mezclas de reacción de Fischer, lo cual ha sido un grave problema en el pasado. Tras algunas mejoras en este método de síntesis, Fischer adoptó posteriormente la síntesis de Koenigs-Knorr para sus investigaciones. Mediante este proceso, E. Fischer y B. Helferich fueron los primeros en reportar la síntesis de un alquilglucósido de cadena larga con propiedades tensioactivas en 1911.
Ya en 1893, Fischer había observado correctamente propiedades esenciales de los alquilglicósidos, como su alta estabilidad a la oxidación y la hidrólisis, especialmente en medios fuertemente alcalinos. Ambas características son valiosas para los alquilpoliglicósidos en aplicaciones tensioactivas.
La investigación relacionada con la reacción de glicosidación continúa y recientemente se han desarrollado varias rutas interesantes para la obtención de glicósidos. Algunos de los procedimientos para la síntesis de glicósidos se resumen en la Figura 5.
En general, los procesos de glicosidación química se pueden dividir en procesos que conducen a equilibrios de oligómeros complejos en el intercambio de glicosilo catalizado por ácido.
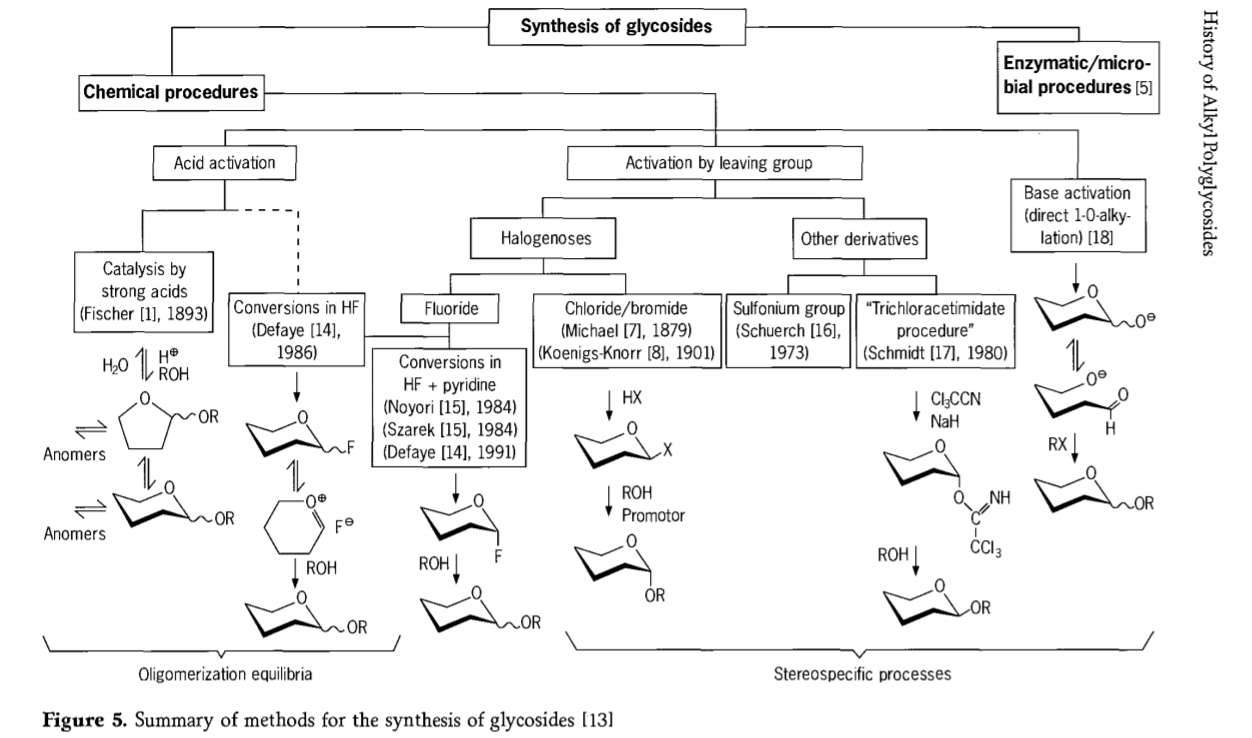
Reacciones sobre sustratos de carbohidratos adecuadamente activados (reacciones glucosídicas de Fischer y reacciones de fluoruro de hidrógeno (HF) con moléculas de carbohidratos desprotegidas) y reacciones de sustitución cinéticamente controladas, irreversibles y principalmente estereotáxicas. Un segundo tipo de procedimiento puede conducir a la formación de especies individuales en lugar de mezclas complejas de reacciones, especialmente al combinarse con técnicas de grupos de conservación. Los carbohidratos pueden dejar grupos en el carbono ectópico, como átomos de halógeno, sulfonilos o grupos tricloroacetimidato, o ser activados por bases antes de su conversión a ésteres triflato.
En el caso particular de las glicosidaciones en fluoruro de hidrógeno o en mezclas de fluoruro de hidrógeno y piridina (polifluoruro de hidrógeno de piridinio), los fluoruros de glicosilo se forman in situ y se convierten sin problemas en glicósidos, por ejemplo, con alcoholes. Se ha demostrado que el fluoruro de hidrógeno es un medio de reacción fuertemente activador y no degradante; se observa autocondensación en equilibrio (oligomerización), similar al proceso de Fischer, aunque el mecanismo de reacción probablemente sea diferente.
Los alquilglicósidos químicamente puros solo son adecuados para aplicaciones muy especiales. Por ejemplo, se han utilizado con éxito en la investigación bioquímica para la cristalización de proteínas de membrana, como la cristalización tridimensional de porina y bacteriorodopsina en presencia de octil β-D-glucopiranósido (experimentos posteriores basados en este trabajo condujeron a la obtención del Premio Nobel de Química para Deisenhofer, Huber y Michel en 1988).
Durante el desarrollo de los alquilpoliglucósidos, se han empleado métodos estereoselectivos a escala de laboratorio para sintetizar diversas sustancias modelo y estudiar sus propiedades fisicoquímicas. Debido a su complejidad, la inestabilidad de los intermediarios y la cantidad y la naturaleza crítica de los residuos del proceso, las síntesis de tipo Koenigs-Knorr y otras técnicas de grupos protectores plantearían importantes problemas técnicos y económicos. Los procesos de tipo Fischer son comparativamente menos complejos y más fáciles de implementar a escala comercial, por lo que son el método preferido para la producción de alquilpoliglucósidos a gran escala.
Hora de publicación: 12 de septiembre de 2020